|
 INTRODUCTION — Movement disorders are
characterized by either reduced (bradykinetic) or
excessive (hyperkinetic) activity. Bradykinetic
movement disorders frequently are accompanied by
rigidity, postural instability, and loss of
automatic associated movements. Diagnosis of the
specific condition depends primarily upon careful
observation of the clinical features. Many such
disorders, mostly rare, exist and only four are
discussed here: Parkinson disease Wilson disease
Huntington disease Hallervorden-Spatz disease
(Neurodegeneration with brain iron accumulation)
INTRODUCTION — Movement disorders are
characterized by either reduced (bradykinetic) or
excessive (hyperkinetic) activity. Bradykinetic
movement disorders frequently are accompanied by
rigidity, postural instability, and loss of
automatic associated movements. Diagnosis of the
specific condition depends primarily upon careful
observation of the clinical features. Many such
disorders, mostly rare, exist and only four are
discussed here: Parkinson disease Wilson disease
Huntington disease Hallervorden-Spatz disease
(Neurodegeneration with brain iron accumulation)
ANATOMY OF THE BASAL GANGLIA — A brief review
of the anatomy of the basal ganglia is appropriate
because this site is involved in many of the
bradykinetic disorders. The basal ganglia regulate
the initiation, scaling, and control of the
amplitude and direction of movement. Movement
disorders can result from biochemical or structural
abnormalities in these structures. The basal ganglia
are a complex of deep nuclei that consist of the
corpus striatum, globus pallidus, and substantia
nigra. The corpus striatum, which includes the
caudate nucleus and the putamen, receives input from
the cerebral cortex and the thalamus and, in turn,
projects to the globus pallidus.
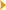 The substantia nigra is divided into the
dopamine-rich pars compacta and the less dense pars
reticularis. The pars reticularis is similar
histologically and chemically to the medial segment
of the globus pallidus, and both project via the
thalamus to the premotor and motor cortex. The
substantia nigra pars compacta gives rise to the
nigral-striatal pathway, which is the main
dopaminergic tract.
The substantia nigra is divided into the
dopamine-rich pars compacta and the less dense pars
reticularis. The pars reticularis is similar
histologically and chemically to the medial segment
of the globus pallidus, and both project via the
thalamus to the premotor and motor cortex. The
substantia nigra pars compacta gives rise to the
nigral-striatal pathway, which is the main
dopaminergic tract.
The output of the basal ganglia projects by way of
the thalamus to the cerebral cortex and then to the
pyramidal system. Basal ganglia output is known as
the extrapyramidal system because it was formerly
thought to be in parallel with the pyramidal system.
Integration of the basal ganglia with the cortex
facilitates motor control.
PARKINSON DISEASE — Bradykinetic movement
disorders consist predominantly of conditions with
features of parkinsonism, of which Parkinson disease
(PD) is the most prominent example. Characteristic
findings include rigidity, akinesia, and gait
disturbance.
Clinical features — PD typically presents in
middle and late life. However, early-onset disease
can occur before age 40 years, and a juvenile form
presents before age 20. Most affected children have
a rigid, akinetic disorder, although many have a
typical resting tremor. Dystonia often involving the
legs, levodopa-induced dyskinesias, and
levodopa-related motor fluctuations (eg, "wearing
off" and "on-off" responses several hours following
a dose) are common in the juvenile form.
Genetics — Most cases of PD are sporadic, but
genetic loci (PARK1 through PARK13) with causative
mutations in six nuclear genes have been associated
with autosomal dominant or recessive Parkinson
disease or parkinsonism. These genes encode the
following proteins: Alpha synuclein Ubiquitin
carboxyl-terminal hydrolase-1 (UCHL1)Parkin DJ1
PTEN-induced putative kinase 1 (PINK1) Leucine
rich repeat kinase 2 (LRRK2), also called dardarin.
Most sporadic cases of PD do not show clear familial
aggregation, but genetic factors likely contribute
to PD susceptibility. One genetic locus (PARK10) on
chromosome 1p has been associated with late onset
idiopathic PD, and mutations in the
glucocerebrosidase gene (GBA) in Ashkenazi Jews have
been associated with a significantly increased risk
of PD compared with healthy controls.
The pathogenesis of autosomal dominant or recessive
Parkinson disease is not completely understood. A
speculative unifying model suggested by genetic
analysis proposes the following mechanisms: Abnormal
aggregation and misfolding of alpha synuclein leads
to Lewy body formation, triggering cellular
oxidative stress and energy depletion. Mutations in
parkin and UCHL1 may interfere with proteosome
degradation of abnormal proteins such as alpha
synuclein. Mutations in DJ1 may enhance misfolding
and aggregation of alpha synuclein. Mutations in DJ1
and PINK1 may contribute to increased oxidative
stress and decreased cellular resistance to stress
imposed by misfolded and abnormally aggregated
proteins. Mutations in GBA may lead to reduced lipid
binding of alpha synuclein and thus an increased
pool available for misfolding and aggregation.
Phenotypic variability — Some have argued
that it is premature to claim that all of these gene
mutations cause true Parkinson's disease, since Lewy
bodies are not clearly associated with either DJ1 or
PINK1 mutations. In addition, there is phenotypic
variability between these different mutations. Alpha
synuclein mutations (PARK1 and PARK4) are associated
with an autosomal dominant inheritance mode; the
phenotype varies from classic Parkinson's disease to
dementia with Lewy bodies. Many patients with
early-onset autosomal recessive familial PD and
isolated juvenile-onset disease have mutations in
the parkin gene (PARK2), located on chromosome
6q25.2-27. In one series, mutations occurred more
frequently in patients with isolated disease when
the age of onset was before 20 than after 30 years
(77 versus 3 percent). In the patients who died,
neuropathologic examination of the brains showed
depigmentation of the substantia nigra pars
compacta. However, the neurons did not contain the
eosinophilic cytoplasmic inclusions (Lewy bodies)
typically seen in PD.
However, the parkin-associated phenotype can be
indistinguishable from idiopathic Parkinson's
disease in some individuals, as evidenced by
detailed evaluation of a large pedigree of parkin
mutation carriers from northern Italy. Among the 77
parkin mutation carriers, 25 had levodopa-responsive
parkinsonism, and five of them met criteria for
definite Parkinson's disease. Neuropathologic
examination of one 73 year old patient who carried
two mutant parkin alleles demonstrated Lewy bodies
in substantia nigra and locus ceruleus. Mutations of
DJ1 (PARK7) are associated with autosomal recessive
inheritance, age younger than 40 at onset, slow
progression, and good response to levodopa.
Mutations of PINK1 (PARK6) are associated with
autosomal recessive inheritance, age younger than 50
at onset, slow progression, and excellent response
to levodopa, similar to parkin and DJ1. PARK6 has
been found worldwide. Although preliminary evidence
suggested that PINK1 was not associated with
sporadic forms of PD, a subsequent report from Italy
found that PINK1 was responsible for some sporadic
cases of early onset PD. The LRRK2 gene (PARK8
locus) maps to chromosome 12p11.2-q13.1 and codes
for dardarin, a protein of unknown function whose
structure suggests it may be a cytoplasmic protein
kinase. Mutations in LRRK2 are associated with
parkinsonism characterized by typical clinical
features of PD, including levodopa responsiveness.
However, the age of onset is highly variable (range
35 to 78 years). Furthermore, the neuropathologic
features may be variable even within the same
family; these include abnormalities consistent with
Lewy body PD, diffuse Lewy body disease, nigral
degeneration without distinctive histopathology, and
tau pathology suggestive of progressive supranuclear
palsy.
The LRRK2 gene may account for a significant
proportion of PD cases. Genetic screening studies
suggest that the Gly2019Ser mutation in the LRRK2
gene accounts for 3 to 13 percent of autosomal
dominant PD in Europe, and 41 percent of autosomal
dominant PD in families from North Africa. The
Gly2019Ser mutation has been identified in
asymptomatic carriers, suggesting reduced or
age-dependent penetrance. LRRK2 mutations have been
found in 0.4 to 1.6 percent of patients with
idiopathic PD, although such cases could also be
explained by reduced penetrance in familial disease.
Diagnosis — The diagnosis of juvenile
parkinsonism is based on clinical signs. Patients
have gradual onset of slowness of movement, tremors
in the hands or legs (but not the head), rigidity of
muscles, shuffling gait, and postural instability.
Other signs include lack of facial expression
(hypomimia), drooling, dysarthria, and dystonia
(involuntary spasms and abnormal postures of hands
and feet).
Treatment — Levodopa is the most effective
drug in the treatment of PD. However, most patients
develop abnormal involuntary movements (dyskinesias)
and unpredictable fluctuations in motor functioning
within three years of treatment. Patients with onset
before age 20 years are most likely to be affected.
As a result, therapy is initiated with other drugs
that will control the symptoms and delay the need
for levodopa. They include anticholinergic drugs
(eg, trihexyphenidyl, amantadine) and dopamine
agonists (eg, pramipexole, ropinirole, and
pergolide).
Complications of levodopa are managed by adjusting
the dosage and frequency of administration. If these
changes do not alleviate symptoms, surgical
treatment, such as high frequency stimulation of the
subthalamic nucleus or globus pallidus, is
considered.
WILSON DISEASE — Wilson disease (WD,
hepatolenticular degeneration) is a treatable cause
of juvenile parkinsonism, dystonia, and other
movement disorders. This rare disorder has an
estimated prevalence of 30 per million. WD is an
autosomal recessive defect of cellular copper
export. The major abnormality in WD is reduced
biliary excretion of copper that leads to its
accumulation, initially in the liver and then in
other tissues, particularly the brain. Tissue copper
deposition causes a multitude of signs and symptoms
that reflect hepatic, neurologic, hematologic, and
renal impairment. The incorporation of copper into
ceruloplasmin is also impaired.
The pathogenesis, diagnosis and treatment of WD are
discussed in detail separately in the appropriate
topic reviews. (See "Pathogenesis and clinical
manifestations of Wilson's disease", see "Diagnosis
of Wilson's disease" and see "Treatment of Wilson's
disease").
HUNTINGTON DISEASE — Huntington disease (HD)
typically presents during the fourth and fifth
decades of life; however, onset occurs during
childhood or adolescence in approximately 5 to 7
percent of affected patients.
Genetics — The genetics and pathogenesis of
HD are discussed in detail separately. (See
"Genetics and pathogenesis of Huntington disease").
Reviewed briefly, HD is transmitted as an autosomal
dominant trait with the affected gene being on the
short arm of chromosome 4. Juvenile onset disease
shows a major transmitting parent effect, as
approximately 80 percent of symptomatic patients
inherit the mutant HD gene from their father. The
high number of cellular divisions that occur during
spermatogenesis likely accounts for the pronounced
paternal-repeat instability.
HD is one of a number of disorders that are
associated with expansion of unstable trinucleotide
(CAG) repeats that encode for polyglutamine tracts
in the protein products. There is mounting evidence
that fragments of the huntingtin protein containing
expanded polyglutamine tracts may be neurotoxic. The
greater the number of CAG repeats on expanded
alleles, the earlier the age of onset and more
severe the disease.
Demonstration of more than 36 CAG repeats in one of
the alleles in the HD gene confirms the diagnosis of
HD. Alleles with 27 to 35 CAG repeats are termed
intermediate alleles. An individual with an allele
in this range is not at risk of developing symptoms
of HD, but may be at risk of having a child with an
allele in the disease range. Juvenile forms are
associated with alleles containing more than 60 to
70 repeats and, in some patients, more than 100
repeats.
The inverse relationship between age of onset and
number of CAG repeats was confirmed in a Dutch
cohort of 755 affected patients. The correlation was
stronger for paternal than maternal inheritance.
Clinical features — Patients with
juvenile-onset HD develop dystonia, ataxia, and
seizures. Most of them have the akinetic-rigid
syndrome termed the Westphal variant. Approximately
one-fourth have the classic feature of chorea seen
in adults. Children also have more rapidly
progressive disease than adults.
Biochemical changes observed in the brains of adults
with HD may explain the neurologic features.
Glutamic acid decarboxylase activity is reduced,
especially in the corpus striatum, substantia nigra,
and other basal ganglia. In contrast,
thyrotropin-releasing hormone, neurotensin,
somatostatin, and neuropeptide Y are increased in
the corpus striatum. The depletion of
gamma-aminobutyric acid in the corpus striatum may
result in disinhibition of the nigral-striatal
pathway. Coupled with the accumulation of
somatostatin, the net result may be the release of
striatal dopamine, which results in chorea.
The pattern of brain abnormality depends upon the
age of onset. The characteristic pathologic change
in adults with HD is diffuse, marked atrophy of the
neostriatum that may be worse in the caudate than in
the putamen. The changes are more dramatic in
early-onset HD. Affected patients typically show
generalized brain atrophy and loss of cerebellar
Purkinje cells.
Treatment — Dopamine-blocking drugs, such as
haloperidol, and dopamine-depleting agents,
including tetrabenazine, often are useful in
controlling chorea. Tetrabenazine is available in
Canada and several other countries.
A randomized controlled trial found that
tetrabenazine at adjusted doses of up to 100 mg
daily was effective for reducing chorea in
ambulatory patients with HD compared with placebo.
However, tetrabenazine treatment was associated with
significantly more adverse events than placebo
treatment. Dose-limiting symptoms with tetrabenazine
included sedation, akathisia, parkinsonism, and
depressed mood; these generally resolved with dosage
adjustments. Two small randomized trials in adults
found that the NMDA-receptor antagonist amantadine
also decreases choreic dyskinesias, however a third
small trial found no such benefit. Levodopa may
provide symptomatic relief of the parkinsonian
features of childhood HD.
Future directions — Many different potential
therapies have shown some promise in animal models
of Huntington disease. These include paroxetine,
coenzyme Q10, minocycline, sodium butyrate,
essential fatty acids, remacemide, creatine,
cystamine, cysteamine and riluzole. Preliminary
clinical trials of many of these agents are
underway.
NEURODEGENERATION WITH BRAIN IRON ACCUMULATION
— Neurodegeneration with brain iron accumulation
(NBIA), formerly known as Hallervorden-Spatz
disease, is a rare progressive neurodegenerative
disorder that causes parkinsonism, dystonia,
cognitive decline, and other neurologic deficits in
children. The onset of NBIA typically is between 4
and 12 years, although it may present as
parkinsonian dementia in adults.
Genetics — Most cases are inherited in an
autosomal recessive pattern, but NBIA also occurs
sporadically, and some phenotypically similar cases
appear to be transmitted as an autosomal dominant.
Many patients with NBIA have mutations in the gene
encoding pantothenate kinase 2 (PANK2), localized to
20p12.3, and are said to have pantothenate
kinase-associated neurodegeneration (PKAN). However,
other gene mutations result in a similar phenotype.
The relationship between genotype and phenotype was
evaluated in a study of 123 patients from 98
families with NBIA. The patients were classified as
having classic disease with early onset and rapid
progression or atypical disease with later onset and
slow progression. All patients with classic disease
and one-third of those with atypical disease had
PANK2 mutations. These patients had the
characteristic appearance of hyperintensity within
the hypointense medial globus pallidus on T2
weighted magnetic resonance images, known as eye of
the tiger, that was not seen in patients without
mutations. Prominent speech-related and psychiatric
symptoms were common in patients with atypical
disease and PANK2 mutations and unusual in those
with atypical disease without the mutations or in
those with classic disease.
Clinical features — Children with NBIA have
posture and gait abnormalities, bradykinesia,
rigidity, and other parkinsonian features, including
tremor. Affected patients also may have hyperkinetic
movement disorders, such as dystonia and
choreoathetosis, as well as progressive dysarthria,
dementia, ataxia, spasticity, seizure disorder,
optic atrophy, and retinitis pigmentosa. In the
large series cited above, abnormalities in classic
NBIA were noted with the following frequencies.
Extrapyramidal signs including dystonia, dysarthria,
rigidity, and choreoathetosis — 98 percent
Retinopathy — 68 percent Cognitive decline — 29
percent Corticospinal tract involvement, including
spasticity, hyperreflexia, and extensor toe signs —
25 percent. Few patients had optic atrophy (3
percent) and none had seizures.
The diagnosis of NBIA is based upon clinical
features. Laboratory studies usually are not
helpful. The disease may be suspected when a MRI
scan shows a central focus of increased T2 signal
intensity surrounded by a zone of decreased signal
in the region of the globus pallidus (eye of the
tiger sign). Scintillation counting after infusion
of radioactive iron (59 Fe) may
demonstrate increased iron uptake in the basal
ganglia.
Increased iron uptake is confirmed by postmortem
examination, which reveals the characteristic
pigmentary degeneration of the basal ganglia,
particularly the internal segment of the globus
pallidus and the zona reticularis of the substantia
nigra. The pigmentary changes result from marked
iron accumulation in these areas.
The mechanism by which basal ganglia iron uptake is
increased in NBIA is not well understood. Systemic
and cerebrospinal fluid iron levels, as well as
plasma ferritin, transferrin, and ceruloplasmin, all
are normal. Furthermore, disorders of systemic iron
overload, such as hemochromatosis, are not
associated with increased brain iron.
Marked neuroaxonal degeneration with the formation
of spheroids is another distinctive pathologic
feature of NBIA. These glycoprotein-containing
axonal swellings have been attributed to abnormal
lipid membrane peroxidation. It may result from
chelation of ferrous iron caused by increased
cysteine (demonstrated in one patient with NBIA),
leading to the accelerated generation of free
hydroxyl radicals in the presence of
non-protein-bound iron.
Treatment — Treatment of NBIA, including iron
chelation with deferoxamine and antioxidant therapy,
is ineffective. Levodopa and anticholinergic drugs
may provide modest relief of parkinsonian symptoms. |