|
 PARKINSON DISEASE BRADYKINETIC VARIANT — Bradykinetic movement
disorders consist predominantly of conditions with
features of parkinsonism, of which Parkinson disease
(PD) is the most prominent example. Characteristic
findings include rigidity, akinesia, and gait
disturbance.
PARKINSON DISEASE BRADYKINETIC VARIANT — Bradykinetic movement
disorders consist predominantly of conditions with
features of parkinsonism, of which Parkinson disease
(PD) is the most prominent example. Characteristic
findings include rigidity, akinesia, and gait
disturbance.
 Clinical features — PD typically presents in
middle and late life. However, early-onset disease
can occur before age 40 years, and a juvenile form
presents before age 20. Most affected children have
a rigid, akinetic disorder, although many have a
typical resting tremor. Dystonia often involving the
legs, levodopa-induced dyskinesias, and
levodopa-related motor fluctuations (eg, "wearing
off" and "on-off" responses several hours following
a dose) are common in the juvenile form.
Clinical features — PD typically presents in
middle and late life. However, early-onset disease
can occur before age 40 years, and a juvenile form
presents before age 20. Most affected children have
a rigid, akinetic disorder, although many have a
typical resting tremor. Dystonia often involving the
legs, levodopa-induced dyskinesias, and
levodopa-related motor fluctuations (eg, "wearing
off" and "on-off" responses several hours following
a dose) are common in the juvenile form.
Genetics — Most cases of PD are sporadic, but
genetic loci (PARK1 through PARK13) with causative
mutations in six nuclear genes have been associated
with autosomal dominant or recessive Parkinson
disease or parkinsonism. These genes encode the
following proteins: Alpha synuclein Ubiquitin
carboxyl-terminal hydrolase-1 (UCHL1)Parkin DJ1
PTEN-induced putative kinase 1 (PINK1) Leucine
rich repeat kinase 2 (LRRK2), also called dardarin.
Most sporadic cases of PD do not show clear familial
aggregation, but genetic factors likely contribute
to PD susceptibility. One genetic locus (PARK10) on
chromosome 1p has been associated with late onset
idiopathic PD, and mutations in the
glucocerebrosidase gene (GBA) in Ashkenazi Jews have
been associated with a significantly increased risk
of PD compared with healthy controls.
The pathogenesis of autosomal dominant or recessive
Parkinson disease is not completely understood. A
speculative unifying model suggested by genetic
analysis proposes the following mechanisms: Abnormal
aggregation and misfolding of alpha synuclein leads
to Lewy body formation, triggering cellular
oxidative stress and energy depletion. Mutations in
parkin and UCHL1 may interfere with proteosome
degradation of abnormal proteins such as alpha
synuclein. Mutations in DJ1 may enhance misfolding
and aggregation of alpha synuclein. Mutations in DJ1
and PINK1 may contribute to increased oxidative
stress and decreased cellular resistance to stress
imposed by misfolded and abnormally aggregated
proteins. Mutations in GBA may lead to reduced lipid
binding of alpha synuclein and thus an increased
pool available for misfolding and aggregation.
Phenotypic variability — Some have argued
that it is premature to claim that all of these gene
mutations cause true Parkinson's disease, since Lewy
bodies are not clearly associated with either DJ1 or
PINK1 mutations. In addition, there is phenotypic
variability between these different mutations. Alpha
synuclein mutations (PARK1 and PARK4) are associated
with an autosomal dominant inheritance mode; the
phenotype varies from classic Parkinson's disease to
dementia with Lewy bodies. Many patients with
early-onset autosomal recessive familial PD and
isolated juvenile-onset disease have mutations in
the parkin gene (PARK2), located on chromosome
6q25.2-27. In one series, mutations occurred more
frequently in patients with isolated disease when
the age of onset was before 20 than after 30 years
(77 versus 3 percent). In the patients who died,
neuropathologic examination of the brains showed
depigmentation of the substantia nigra pars
compacta. However, the neurons did not contain the
eosinophilic cytoplasmic inclusions (Lewy bodies)
typically seen in PD.
However, the parkin-associated phenotype can be
indistinguishable from idiopathic Parkinson's
disease in some individuals, as evidenced by
detailed evaluation of a large pedigree of parkin
mutation carriers from northern Italy. Among the 77
parkin mutation carriers, 25 had levodopa-responsive
parkinsonism, and five of them met criteria for
definite Parkinson's disease. Neuropathologic
examination of one 73 year old patient who carried
two mutant parkin alleles demonstrated Lewy bodies
in substantia nigra and locus ceruleus. Mutations of
DJ1 (PARK7) are associated with autosomal recessive
inheritance, age younger than 40 at onset, slow
progression, and good response to levodopa.
Mutations of PINK1 (PARK6) are associated with
autosomal recessive inheritance, age younger than 50
at onset, slow progression, and excellent response
to levodopa, similar to parkin and DJ1. PARK6 has
been found worldwide. Although preliminary evidence
suggested that PINK1 was not associated with
sporadic forms of PD, a subsequent report from Italy
found that PINK1 was responsible for some sporadic
cases of early onset PD. The LRRK2 gene (PARK8
locus) maps to chromosome 12p11.2-q13.1 and codes
for dardarin, a protein of unknown function whose
structure suggests it may be a cytoplasmic protein
kinase. Mutations in LRRK2 are associated with
parkinsonism characterized by typical clinical
features of PD, including levodopa responsiveness.
However, the age of onset is highly variable (range
35 to 78 years). Furthermore, the neuropathologic
features may be variable even within the same
family; these include abnormalities consistent with
Lewy body PD, diffuse Lewy body disease, nigral
degeneration without distinctive histopathology, and
tau pathology suggestive of progressive supranuclear
palsy.
The LRRK2 gene may account for a significant
proportion of PD cases. Genetic screening studies
suggest that the Gly2019Ser mutation in the LRRK2
gene accounts for 3 to 13 percent of autosomal
dominant PD in Europe, and 41 percent of autosomal
dominant PD in families from North Africa. The
Gly2019Ser mutation has been identified in
asymptomatic carriers, suggesting reduced or
age-dependent penetrance. LRRK2 mutations have been
found in 0.4 to 1.6 percent of patients with
idiopathic PD, although such cases could also be
explained by reduced penetrance in familial disease.
Diagnosis — The diagnosis of juvenile
parkinsonism is based on clinical signs. Patients
have gradual onset of slowness of movement, tremors
in the hands or legs (but not the head), rigidity of
muscles, shuffling gait, and postural instability.
Other signs include lack of facial expression
(hypomimia), drooling, dysarthria, and dystonia
(involuntary spasms and abnormal postures of hands
and feet).
Treatment — Levodopa is the most effective
drug in the treatment of PD. However, most patients
develop abnormal involuntary movements (dyskinesias)
and unpredictable fluctuations in motor functioning
within three years of treatment. Patients with onset
before age 20 years are most likely to be affected.
As a result, therapy is initiated with other drugs
that will control the symptoms and delay the need
for levodopa. They include anticholinergic drugs
(eg, trihexyphenidyl, amantadine) and dopamine
agonists (eg, pramipexole, ropinirole, and
pergolide).
Complications of levodopa are managed by adjusting
the dosage and frequency of administration. If these
changes do not alleviate symptoms, surgical
treatment, such as high frequency stimulation of the
subthalamic nucleus or globus pallidus, is
considered.
 Motor fluctuations and dyskinesia in Parkinson's
disease
Motor fluctuations and dyskinesia in Parkinson's
disease
INTRODUCTION — As many as 50 percent of patients
on levodopa for five years experience motor
fluctuations (MF) and dyskinesia. These symptoms are
especially common in patients with young-onset (eg,
under the age of 50) Parkinson's disease (PD); they
are unique to levodopa and are not produced by the
other antiparkinson drugs.
Patients typically experience a smooth and even
response to the early stages of levodopa treatment.
As the disease advances, however, the effect of
levodopa begins to wear off approximately four hours
after each dose, leaving patients anticipating the
need for their next dose. This phenomenon may be
explained by the observation that dopamine nerve
terminals are able to store and release dopamine
early in the course of disease but, with more
advanced disease and increasing degeneration of
dopamine terminals, the concentration of dopamine in
the basal ganglia is much more dependent upon plasma
levodopa levels. Plasma levels may fluctuate
erratically because of the 90 minute half-life of
levodopa and the frequently unpredictable intestinal
absorption of this medication.
Motor fluctuations (MF) are alterations between
periods of being "on," during which the patient
enjoys a good response to medication, and being
"off" during which the patient experiences symptoms
of their underlying parkinsonism.
Dyskinesia consists of abnormal involuntary
movements that are usually choreic or dystonic but,
when more severe, may be ballistic or myoclonic.
Dyskinesia usually appears when the patient is "on."
It may occasionally occur in the form of painful
dystonia when the patient is "off," especially in
the morning on awakening, when dystonic intorsion of
a foot (usually on the side of greater parkinsonian
involvement) occurs as a withdrawal reaction because
of the long interval without medication overnight.
Surgery for advanced PD is another therapeutic
option, as bilateral deep brain stimulation of the
subthalamic nucleus or globus pallidus appears to
improve motor function in selected patients with
advanced typical PD and MF, whose condition cannot
be further improved by medical therapy. The surgical
management of advanced PD is discussed
elsewhere.
WEARING-OFF PHENOMENON — Patients with advanced
Parkinson's disease (PD) begin to be aware of a
wearing "off" or end-of-dose effect less than four
hours following a dose of levodopa.
Alteration of levodopa dosing — Wearing "off" can
initially be managed by increasing the dose of
levodopa, if the patient is not having side effects
and is taking a relatively low dose. However,
increasing the dose often increases side effects
without effectively increasing the dose duration.
Shortening the interdose interval while
administering lower doses is usually a more
effective approach. However, it is often difficult
to titrate the dose precisely, and some patients
begin to exhibit an "all or none" response whereby
individual lower doses produce no evident clinical
response. This occurs because the pharmacologic
response threshold is higher in advanced disease
than it is in earlier disease.
Liquid Sinemet (carbidopa/levodopa) is occasionally
used for patients when titration of the dose and
dose interval using tablets is difficult. However,
this approach is not typically practical since
Sinemet is insoluble in water and no commercial
preparation of liquid Sinemet is available.
Instructions for preparation of a daily supply of
liquid Sinemet are available, but use of this
approach is best left to the specialist.
The sustained-release forms of levodopa preparations
(eg, Sinemet CR) may be useful in the early stages
of the wearing "off" phenomenon and may add up to 90
additional minutes throughout the day to levodopa's
duration of effect. However, Sinemet CR is less
well absorbed than immediate release Sinemet; thus,
an individual dose increase of approximately 30
percent may be required to achieve the same clinical
response.
A
practice parameter from the American Academy of
Neurology (AAN) issued in 2006 concluded that
sustained-release carbidopa/levodopa does not
decrease "off" time compared with immediate release
formulations.
Addition of a second drug — Addition of a second
drug is indicated if the adjustments cited above are
not successful.
1. Dopamine agonists — Dopamine agonists are commonly
used to reduce the amount of "off" time in patients
with advanced PD and may also allow for the dose of
levodopa to be reduced. The drugs currently
approved by the United States Food and Drug
Administration (FDA) include bromocriptine
(Parlodel), pergolide (Permax), pramipexole
(Mirapex), ropinirole (Requip), and apomorphine
(Apokyn). Cabergoline is approved by the FDA only
for the treatment of hyperprolactinemic disorders,
and its use for advanced PD is off label.
Studies comparing the efficacy of various dopamine
agonists have found either no significant difference
or only mild superiority of one agent over another.
The dopamine agonist apomorphine administered
subcutaneously can be used for rapid onset (usually
within 10 minutes) rescue therapy when patients
suddenly turn "off". In a randomized,
double-blind, placebo-controlled study of 29
patients with advanced PD and two hours or more of
"off" time despite aggressive oral therapy,
administration of subcutaneous apomorphine (2 to 10
mg) resulted in successful amelioration of "off"
state events following 95 percent of injections
compared with 23 percent receiving placebo injection.
One review concluded that the magnitude and pattern
of the motor response to a single subcutaneous dose
of apomorphine is qualitatively comparable to that
of oral levodopa; a 4 mg dose achieved a clinically
significant improvement in 75 percent of patients.
Cabergoline may have some utility for reduction of
"off" time in patients with advanced PD, but data
are limited. In a single center, 24-week study of 37
patients (19 active, 18 placebo), treatment with
cabergoline (mean dose 5.4 mg/day) was associated
with a significant decrease in daily "off" time
compared with placebo (2 versus 0.7 hours [40 versus
18 percent]). However, these results are
limited by a potentially confounding baseline
difference in "off" time duration between the
treatment groups. In another single center,
24-week study of 27 patients (17 active, 10
placebo), patients treated with cabergoline (mean
dose 4.9 mg/day) had an increase in "on" time (2.7
hours [30 percent]) and a decrease in "off" time
(3.3 hours [59 percent]), but no information
was provided for the placebo group about these
parameters.
Cabergoline treatment was not associated with
increased dyskinesia in these trials.
However, a retrospective case control study of 210
patients with PD found that high cumulative dose and
long-term treatment with cabergoline was associated
with an increased risk of cardiac valvulopathy
detected on transthoracic echocardiography.
In a randomized controlled trial, bromocriptine
decreased "off" time compared with placebo (8 versus
3 percent, respectively), but the difference was not
statistically significant.
2. COMT inhibitors — Catechol-O-methyl transferase
(COMT) inhibitors such as tolcapone (Tasmar) and
entacapone (Comtan) may prolong and potentiate the
levodopa effect and reduce the "off" time when given
with a dose of levodopa. The net result is
an increased levodopa effect in fluctuating patients.
These medications may allow a reduction in the total
daily levodopa dose by as much as 30 percent.
The starting dose of tolcapone is 100 mg three times
daily; the clinical effect is evident immediately.
The dose of entacapone is one 200 mg tablet with
each dose of levodopa, up to a maximum of eight
doses per day.
The most common side effects of these drugs are due
to increased dopaminergic stimulation and include
dyskinesia, psychiatric effects (mainly visual
hallucinations), nausea, diarrhea, and orthostatic
hypotension. The adverse effects are managed by
lowering the dose of levodopa either before or after
the addition of tolcapone or entacapone. Both drugs
may also cause a brown-orange urine discoloration.
In clinical trials, tolcapone was associated with
transient, asymptomatic elevations of transaminases
(AST and ALT) in 1 to 3 percent of subjects exposed
to the drug. Three reported deaths from
hepatotoxicity in patients using tolcapone prompted
its removal from the market in Canada and Europe, although it is still available in the United
States with the recommendation that it be used for
treatment of motor fluctuations only after other
methods have been exhausted and with monitoring of
ALT and AST levels for the first six months of
therapy. Entacapone has thus far not been associated
with hepatotoxicity.
Monitoring of liver enzymes with liver function
tests (LFTs) must be done at baseline and then every
two weeks for the first year of tolcapone therapy,
then every four weeks for the next six months, then
every eight weeks thereafter. Monitoring of LFTs
should be resumed at the previous frequency if the
tolcapone dose is increased to 200 mg three times a
day. Tolcapone should be discontinued if the ALT or
AST exceeds the upper limit of normal or if the
clinical signs and symptoms suggest the onset of
liver failure.
3. MAO B inhibitors —
Rasagiline is a selective
monoamine oxidase (MAO) B inhibitor. It has
potential long-term effects on dopamine transmission
because it acts irreversibly on MAO B receptors.
Rasagiline appears to be effective for motor
complications in PD as demonstrated in randomized
clinical trials. One of these, the 18-week
multicenter LARGO trial, evaluated 687 patients with
PD who had motor fluctuations (MF) for at least one
hour every day despite optimum levodopa/dopa
decarboxylase therapy. Patients were randomly
assigned to adjunct therapy with either rasagiline 1
mg daily, entacapone 200 mg with every levodopa
dose, or placebo. Both rasagiline and entacapone
reduced mean daily "off" time (the primary outcome
measure) by about one hour compared with placebo,
and both increased daily "on" time without
troublesome dyskinesia compared with placebo. The
beneficial effect of rasagiline was independent of
age (<70 versus 70 years) and of adjunct use of
dopamine agonists.
Rasagiline was well tolerated in these studies. The frequency of dopaminergic adverse
events in the LARGO trial was similar to that seen
in the entacapone and placebo groups.
Rasagiline is approved by the European Commission as
initial monotherapy in patients with early PD and as
adjunct treatment in moderate to advanced PD. It
received similar approval by the United States Food
and Drug Administration in May 2006.
Selegiline is another selective MAO B inhibitor.
Unlike rasagiline, selegiline is metabolized to
amphetamine derivatives. Although selegiline may
extend the levodopa effect, the clinical benefit
this produces is usually relatively mild.
Results from a small randomized controlled trial
suggest that orally disintegrating selegiline may
also be beneficial, although the study did not
report change in levodopa dose.
Other strategies — Anticholinergic drugs and
amantadine are not very effective in managing the
wearing "off" effect and are rarely indicated for
this purpose, given the other more effective
options.
Early studies of adenosine A2A antagonists as
adjunctive therapy in PD have yielded promising
results, and clinical trials are ongoing.
Preliminary studies suggest that eradication of
Helicobacter colonization, which is present in about
half of the population, may be a useful method for
improving levodopa absorption and reducing motor
fluctuations in patients with PD. These
require confirmation in larger clinical trials
before routine testing for H. pylori and antibiotic
eradication can be recommended.
Guideline recommendations for treating "off" time —
An evidenced-based practice parameter from the AAN
issued in 2006 made the following recommendations
for the treatment of "off" time in patients with PD
and motor fluctuations: Entacapone and rasagiline are established as effective and
should be offered to reduce "off" time. Pergolide,
pramipexole, ropinirole, and tolcapone are probably effective and should be considered to
reduce "off" time, with the stipulation that the
adverse effects of tolcapone (hepatotoxicity) and
pergolide (valvular fibrosis) require that they be
used with caution and monitoring. Apomorphine,
cabergoline, and
selegiline are
possibly effective and may be considered to reduce
"off" time. Sustained release carbidopa/levodopa does not
decrease "off" time compared with immediate release
carbidopa/levodopa; bromocriptine does not reduce "off" time compared
with placebo; both may be disregarded to reduce
"off" time.
UNPREDICTABLE OFF PERIODS — Transitions from
being "on" to being "off" can be sudden and
unpredictable in some patients. Unlike the wearing
"off" phenomenon at the end of a levodopa dose
cycle, there is sometimes no obvious relationship
between the time of levodopa administration and the
appearance of "off" episodes in patients with
unpredictable "off" periods. These periods typically
occur in patients with advanced Parkinson's disease
(PD) who are also experiencing motor fluctuations
(MF) and severe dyskinesia.
Management of these individuals is similar to that
for patients who are having problems with wearing
"off," although it typically is much more difficult.
Direct observation of the patient during a prolonged
outpatient visit as he or she cycles through such
episodes is useful to determine the relationship of
levodopa doses to "off" episodes. In some cases,
these episodes occur at times of peak levodopa
effect due to excessive rather than insufficient
dopaminergic stimulation; such patients are best
treated by reducing rather than raising the levodopa
dose.
Addition of a COMT inhibitor or a dopamine agonist
can be helpful; marked reduction of the levodopa
dose together with the addition of high doses of a
dopamine agonist may be required. Controlled release
levodopa preparations (eg, Sinemet CR) are usually
not helpful and occasionally exacerbate the
situation.
Competition with neutral amino acids for transport
across the gut and into the brain may be responsible
for "offs" that appear following meals. A
protein redistribution diet in which most protein
intake is reserved for the evening was useful in
approximately two-thirds of such patients in small
studies, although this type of diet tends to
be impractical for long-term use.
Episodic freezing is a special form of unpredictable
"off" in which patients suddenly become immobilized
for seconds to minutes at a time. This complication
usually occurs while walking when it may cause
falls; it is often not medication related and is
very resistant to treatment. When freezing is more
prolonged and accompanied by the emergence of other
parkinsonian signs, treatment is similar to patients
with other forms of the wearing "off" effect.
ACUTE AKINESIA — Acute akinesia is a sudden
exacerbation of Parkinson's disease (PD)
characterized by an akinetic state that lasts for
several days and does not respond to treatment with
antiparkinson medication. This phenomenon is
different from wearing "off" effects and may occur in
patients not previously treated with levodopa.
Acute akinesia should prompt a search for systemic
infection or other intercurrent medical problems
that are capable of causing a sudden worsening of
parkinsonism. In a review of this problem in 26
patients, acute akinesia appeared after a flu-like
syndrome in six patients, hip joint surgery or bone
fractures in eight patients, gastrointestinal
disturbances in three patients, and various
medication manipulations in the remaining patients. Four patients died in spite of treatment.
Episodes of acute akinesia may therefore have
serious consequences and usually warrant acute
hospitalization in order to identify and correct the
underlying cause.
FAILURE OF ON-RESPONSE — Patients with motor
fluctuations (MF) sometimes fail to turn "on"
following a dose of levodopa. This has been called
the "no-on" response. In some cases, this
is due to delayed gastric motility, which results in
inadequate plasma concentrations in advanced
patients who have a narrow therapeutic window. A
common reason for the "no-on" phenomenon is an
excessively prolonged or severe "off" period
occurring before the "no-on." This is best managed
by avoiding "offs."
The prokinetic agent cisapride increases
gastrointestinal motility and may be helpful in such
patients, but the drug has been associated with a
number of drug interactions and fatal cardiac
arrhythmias, prompting the manufacturer to severely
restrict its availability in the United States. The
prokinetic drug metoclopramide is a dopamine
receptor blocker that should be avoided. Patients
should be encouraged to take levodopa on an empty
stomach and avoid protein at the time of drug
administration.
Domperidone is a D2-blocker with selective
peripheral activity in the upper gastrointestinal
tract; it does not cross the blood-brain barrier and
therefore lacks the neurologic side effects of
metoclopramide. It is currently not available in the
United States but is available in Canada and other
countries. Although data are limited, domperidone
(starting at 20 mg four times daily) may be useful
as a prokinetic agent to treat delayed gastric
emptying in patients with PD. However,
animal studies suggest that, like cisapride,
domperidone may increase the risk of cardiac
arrhythmias.
DYSKINESIA — Dyskinesia refers to a variety of
involuntary movements, which occur as a direct
effect of levodopa. Other antiparkinson drugs
are much less likely to produce these motor
abnormalities but may exacerbate them once they have
appeared following treatment with levodopa.
Dyskinesia is sometimes mistaken for manifestations
of progressive Parkinson's disease (PD) or confused
with tremor by patients and their families, rather
than recognized as reversible consequences of
treatment.
Dyskinesia occurs in 30 to 40 percent of patients
treated with levodopa by five years and nearly 60
percent by ten years, but not all dyskinesia
requires treatment. A retrospective study suggests
that the rate of dyskinesia requiring medication
adjustment at five and ten years after levodopa
treatment is 17 and 43 percent, respectively.
Dyskinesia is usually choreiform in type, manifested
by continuous, restless appearing movements of the
extremities, head, face, trunk, and respiratory
muscles. These dyskinetic movements are remarkably
well tolerated by most patients since patients feel
entirely relieved of their parkinsonism while
dyskinesia is present. However, severe dyskinesia
may take the form of large amplitude, ballistic
movements that interfere with function and become
very disturbing to patients and their families.
Levodopa was given in relatively high doses when it
was first used as therapy for PD. As a result,
dyskinesia was often seen early in treatment,
especially in those with advanced disease who were
being treated for the first time. The subsequent use
of more modest doses resulted in its later
appearance, months to years after initiating
levodopa. Dyskinesia is especially common in
patients with young-onset PD.
Peak-dose dyskinesia is most common. It occurs
60 to 90 minutes following a dose of levodopa. Early
in the disease, this complication can be managed by
lowering the medication dose, switching to a
controlled release preparation, or reducing
adjunctive drugs such as dopamine agonists,
selegiline, or anticholinergic drugs. However, in
more advanced patients with brittle responses,
reducing the dose of levodopa may result in complete
failure to generate an "on" response. In this
situation, the dose of dopamine agonist should be
greatly increased and the levodopa dose reduced,
since dopamine agonists are much less likely to
induce dyskinesia than levodopa.
An unusual pattern sometimes evolves in which
dyskinesia peaks twice after each dose (diphasic
dyskinesia) - when patients turn "on" and again as
they begin to turn "off". In the second phase,
dyskinesia in one body part may coexist with the
emergence elsewhere of parkinsonian signs such as
tremor and dyskinesia. This pattern is often
unrecognized and may only be appreciated if the
patient is observed during a prolonged outpatient
visit.
The diphasic pattern is notoriously difficult to
manage and usually requires more frequent levodopa
dosing to prevent wearing "off" prior to each dose.
However, this strategy often leads to progressively
increasing dyskinesia as the day goes on. Addition
of a dopamine agonist and a marked reduction in the
levodopa dose should be tried in such patients.
Sustained release levodopa is best avoided in
patients with severe or complex patterns of
dyskinesia since absorption may be delayed and
dyskinesia tends to progressively increase into the
afternoon and evening.
Amantadine — Amantadine may be useful for treating
dyskinesia in advanced PD. Several studies have
shown short-term benefit. As an example, a
single-center randomized controlled trial found that
amantadine administration compared with placebo was
associated with a 24 percent reduction in the total
dyskinesia score. In addition, a
placebo-controlled study involving 17 patients
showed that the beneficial effects of amantadine on
motor response fluctuations were maintained for at
least one year; initial and one-year reductions in
dyskinesia scores were 60 and 56 percent,
respectively.
In another placebo-controlled study in 40 patients
with dyskinesia, amantadine treatment for 15 days
resulted in a 45 percent reduction in dyskinesia
scores compared with placebo. However, the
benefit in this study lasted less than eight months,
and amantadine withdrawal resulted in a rebound with
increase of dyskinesia in 11 patients.
Amantadine was not associated with worsening of
parkinsonism symptoms in these studies.
The dose of amantadine for dyskinesia is one tablet
(100 mg) one to three times a day. Side effects may
include peripheral edema, psychosis, and livedo
reticularis.
Clozapine — Low doses of the antipsychotic clozapine
(30 to 50 mg/day) reduced dyskinesia in several
open-label studies, and low dose clozapine
(12.5 to 75 mg/day) was significantly more effective
than placebo in treating levodopa-induced dyskinesia
in a double-blind, randomized controlled trial of 50
patients. The usefulness of clozapine is
limited by its potential for inducing bone marrow
suppression, but this risk may be acceptably low
with monitoring. Clozapine treatment requires
obtaining the white blood cell count (WBC) and
absolute neutrophil count (ANC) at baseline and
weekly for the first six months of continuous
treatment, followed by biweekly monitoring
thereafter.
The dibenzodiazepine derivative
olanzapine has
similar properties to clozapine. In a randomized
controlled trial of 10 patients with PD, low-dose
olanzapine was effective in reducing dyskinesia, but
was associated with unacceptable increases in
parkinsonism and "off" time.
Guideline recommendations for reducing dyskinesia —
An evidenced-based practice parameter from the AAN
issued in 2006 made the following recommendations
for the treatment of dyskinesia in patients with PD
and motor fluctuations: Amantadine is possibly
effective and may be considered for reducing
dyskinesia. There is insufficient evidence to
support or refute the effectiveness of clozapine in
reducing dyskinesia.
DYSTONIA — Dystonia is a more sustained abnormal
posture than dyskinesia. Dystonic postures usually
involve the limbs but can affect the face, neck, or
trunk.
Dystonia can be a manifestation of early untreated
Parkinson's disease (PD) or may appear as a
complication of levodopa treatment. A careful
history is required since, when due to levodopa,
dystonia can occur either as a peak levodopa effect
or during "off" periods due to levodopa withdrawal.
Withdrawal dystonia most commonly occurs in the
early morning when it produces painful flexion and
inversion postures of the feet and toes.
Peak dystonia is managed similarly to peak
dyskinesia. "Off" period dystonia that occurs early
in the morning is managed either by taking sustained
release levodopa before retiring or by taking
levodopa or a dopamine agonist during the night or
first thing in the morning before arising. "Off"
period dystonia during the day is managed similarly
to other forms of the wearing "off" effect.
Another form of levodopa withdrawal is akathisia
(motor restlessness) or restless legs, which usually
occurs at night, several hours after the last dose
of levodopa. This is managed by providing levodopa
or a dopamine agonist before retiring.
RECOMMENDATIONS
— The following treatment
suggestions represent my approach to some difficult
management issues that occur in advanced Parkinson's
disease (PD).
Wearing "off" phenomenon Document the pattern of
motor fluctuations (MF). Obtain a careful and
accurate history, and observe the patient directly
in an outpatient setting. Examine the effect of
diet, and avoid taking levodopa with high protein
meals. A sustained-release levodopa formulation may
be beneficial, but only in the early stages of
wearing "off" in patients with less advanced PD.
In
patients with more advanced PD, reduce the levodopa
dose interval by 30 to 60 minutes. This may require
the addition of an extra levodopa dose at the end of
the day. In most cases, individual levodopa doses
should be left unchanged. Consider adding the COMT
inhibitors entacapone (Comtan) or tolcapone
(Tasmar). Entacapone should be given first because
of the small risk that tolcapone can cause an
elevation of liver enzymes. Be prepared to lower the
levodopa dose by up to 30 percent because of the
increased peak levodopa effect if tolcapone is used.
Consider adding an
oral dopamine agonist such as pramipexole or
ropinirole. Watch
for dopaminergic toxicity such as visual
hallucinations and confusion, and be prepared to
lower the levodopa dose. Consider parenteral
apomorphine in patients with sudden and severe
wearing "off" effects. This rescue therapy is very
effective, but it has the disadvantage of requiring
a prophylactic antiemetic such as trimethobenzamide.
In addition, the effective dose of parenteral
apomorphine must be established for each patient by
administration during a prolonged outpatient
evaluation prior to initiating therapy. Consider the
MAO B inhibitors rasagiline and selegiline. Be aware that selegiline
exerts only a mild effect on the wearing "off"
phenomenon, while rasagiline has an effect
comparable to entacapone. Rasagiline is now approved
in the United States and in the European Union.
Unpredictable "off" periods Document that "off"
periods are unpredictable and long lasting. In many
cases, they are sudden wearing "off" effects or
transient freezing episodes. Avoid taking levodopa
with high protein meals. Evaluate and treat the
possible effects of anxiety, which may precipitate
sudden "off" episodes. Consider raising the levodopa
dose. Plasma levodopa levels may be falling below
the therapeutic threshold. Alternatively, consider
lowering the levodopa dose. In rare cases, sudden
"off" episodes may be due to excessive levodopa
effects
Failure or delay of the "on" response Avoid taking
levodopa with high protein meals. Examine
gastrointestinal absorption. Avoid wearing "off"
effects. Failure or delay of the "on" responses
often occur after prolonged wearing "off" episodes
Dyskinesia and dystonia:
Lower the levodopa dose when
possible.
Replace a portion of the levodopa dose with
a dopamine agonist, if necessary.
Replace
sustained-release levodopa with regular levodopa, if
dyskinesia is occurring in the late afternoon and
evening.
Add amantadine to counteract dyskinesia.
Manage diphasic dyskinesia with
more frequent levodopa dosing.
Use
middle-of-the-night levodopa or a dopamine agonist
to treat early morning "off" period dystonia.
Reduce
the levodopa dose intervals or add a dopamine
agonist to treat "off" period dystonia during the
day
MRI and MRS in Parkinson disease.
Parkinson’s disease (PD) is a progressive
neurological disorder characterized by a variable
degree of impairment in motor skills, speech, and
other CNS functions. Rest tremor, bradykinesia,
rigidity, and loss of postural reflexes are
generally considered the cardinal signs of PD. Other
clinical features include secondary motor symptoms
(e.g. dysphagia, sialorrhoea, micrographia,
shuffling gait, and festination) and non-motor
symptoms (e.g. autonomic dysfunction,
cognitive/neurobehavioral abnormalities, sleep
disorders, and sensory abnormalities). The symptoms
are the results of decreased stimulation of the
motor cortex by the basal ganglia, normally caused
by the insufficient formation and action of dopamine
due to an idiopathic degeneration of the brain
dopaminergic system. The mechanism by which brain
cells are lost may consist of an abnormal protein
accumulation (alphasynuclein to ubiquitin) in the
damaged cells, which leads to the accumulation of
the characteristic inclusions called Lewy bodies.
Excessive accumulation of iron, which is toxic to
nerve cells, are also typically observed in
conjunction with the protein inclusions. Recently,
genetic mutations, protein mishandling, increased
oxidative stress, mitochondrial dysfunction,
inflammation, and other pathogenic mechanisms have
been identified as contributing factors in the death
of dopaminergic and non-dopaminergic cells in the
brains of PD patients. There are no definitive
diagnostic tests for the diagnosis of PD. Thus the
disease must be diagnosed based on clinical
criteria, which are typically based on the presence
of a combination of cardinal motor features,
associated and exclusionary symptoms, and response
to levodopa. Pathological confirmation of the
hallmark Lewy bodies on autopsy is still considered
the criterion for definite PD diagnosis.
The disease is not fatal, but it progresses with
time, dramatically worsening the subject’s quality
of life and decreasing his/her average life
expectancy.
The treatment includes drug therapy (e.g. levodopa,
dopamine agonists, and monoamine oxidase-B
inhibitors), as well as surgery and deep brain
stimulation (in advanced PD patients for whom drug
therapy is no longer sufficient).
Although the diagnosis of PD is straightforward when
patients have a classical presentation,
differentiating PD from other forms of PD related
disorders is difficult. These affections include
secondary (acquired) Parkinsonism,
progressive supranuclear palsy (PSP),
multiple system degeneration
(MSA), and corticobasal
degeneration (CBD). The absence of rest
tremor, early occurrence of gait difficulty,
postural instability, dementia, and the presence of
dysautonomia, ophthalmoparesis, ataxia, and other
atypical features, coupled with poor or no response
to levodopa, can help in the differential diagnosis
of these disorders.
However, at early disease stages, when signs and
symptoms overlap, this can be very challenging,
leading to a significant number of misdiagnoses. Due
to the very different natural histories of these
diseases, an early differentiation between PD and
related disorders is important for correct prognosis
and treatment strategy.
Conventional MRI is normal in PD patients and
this investigation is usually performed to exclude a
structural cause for the development of
Parkinsonism. At late disease stages, the atrophy of
the substantia nigra may become evident.
Conventional MRI, however, may be useful in the
differentiation of the various Parkinsonian
syndromes, as it frequently shows abnormalities in
these patients. Qualitative and quantitative studies
have shown that atrophy and signal changes in
putamen and infratentorial structures can
differentiate patients with MSA from PD patients
with high specificity at late stages, although
sensitivity is suboptimal especially at early
stages. An MR index based on the midbrain area and
the width of superior cerebellar peduncles was able
to distinguish patients with PSP from those with PD
and MSA with high sensitivity and specificity. On
CBD, the midbrain is generally not atrophic, which
may help to distinguish this condition from other
atypical Parkinsonisms. In some cases, an
asymmetrical atrophy (more marked on the side
opposite to the clinically involved side of the body
and prevalently involving the frontoparietal cortex)
can be found.
1H-MRS
Studies of MRS in PD have produced conflicting
results, showing either no difference in the basal
ganglia metabolite levels between PD patients and
normal controls, or decreases of NAA/Cr levels in
PD. The variability of the results is probably
related to the difficulty of reliably assessing
metabolic abnormalities in the substantia nigra, due
to its small size and high iron content.
Interestingly, by using high field MR strength (4
T), recent work has shown the ability to measure
multiple metabolites (including GABA) very
accurately in a small volume (2.2 ml) including the
substantia nigra, but did not find any differences
between patients and controls in a relatively small
study (10 PD, 11 controls) (Figure-1).[1]
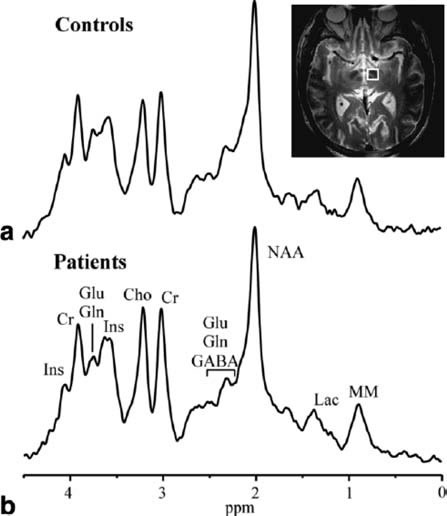
Figure-1: Short TE MRS TE 5 ms, TR 4.5 sec,
400 averages for each subject acquired with 4 T
magnet from 2.2 ml
volumes, that encompass the substantia nigra of (a)
11 healthy volunteers and (b) 10 patients with PD.
The VOI is shown in the T2-weighted images (From Oz
et al. with permission).
In contrast to patients with PD, the reduction of
NAA in basal ganglia and other brain regions seems
to be consistent in patients with related
Parkinsonian disorders. Decreases in basal ganglia
levels of NAA have been reported in patients with
MSA. In an MRS study at high field strength (3 T),
NAA/Cr decreases were confirmed to be significantly
reduced in the putamen and in the pontine base of
MSA patients, suggesting that these measurements may
be of diagnostic value early in the disease course.
Another study was performed in groups of patients
with PD, PSP, and CBD using a multi-slice MRSI
approach, which allowed the assessment of the
metabolic profile of several brain regions with good
spatial resolution. Decreases of NAA/Cr were
observed in PSP patients in brainstem, centrum
semiovale, frontal lobe, and precentral cortex, as
well as a reduction of NAA/Cho in the lentiform
nucleus. However, the largest decrease in NAA/Cho
was in the lentiform nucleus of CBD patients,
exactly where one would expect the most prominent
neuropathological abnormality in this disease.
Again, this study confirmed that the PD patient
group showed no metabolic abnormalities in any of
the brain regions studied.
Collectively, these studies suggest that there is a
potential role for the use of 1H-MRS in the
differential diagnosis of Parkinsonian related
syndromes, and perhaps also for monitoring the
effects of treatment in these disorders.
Case Presentation:
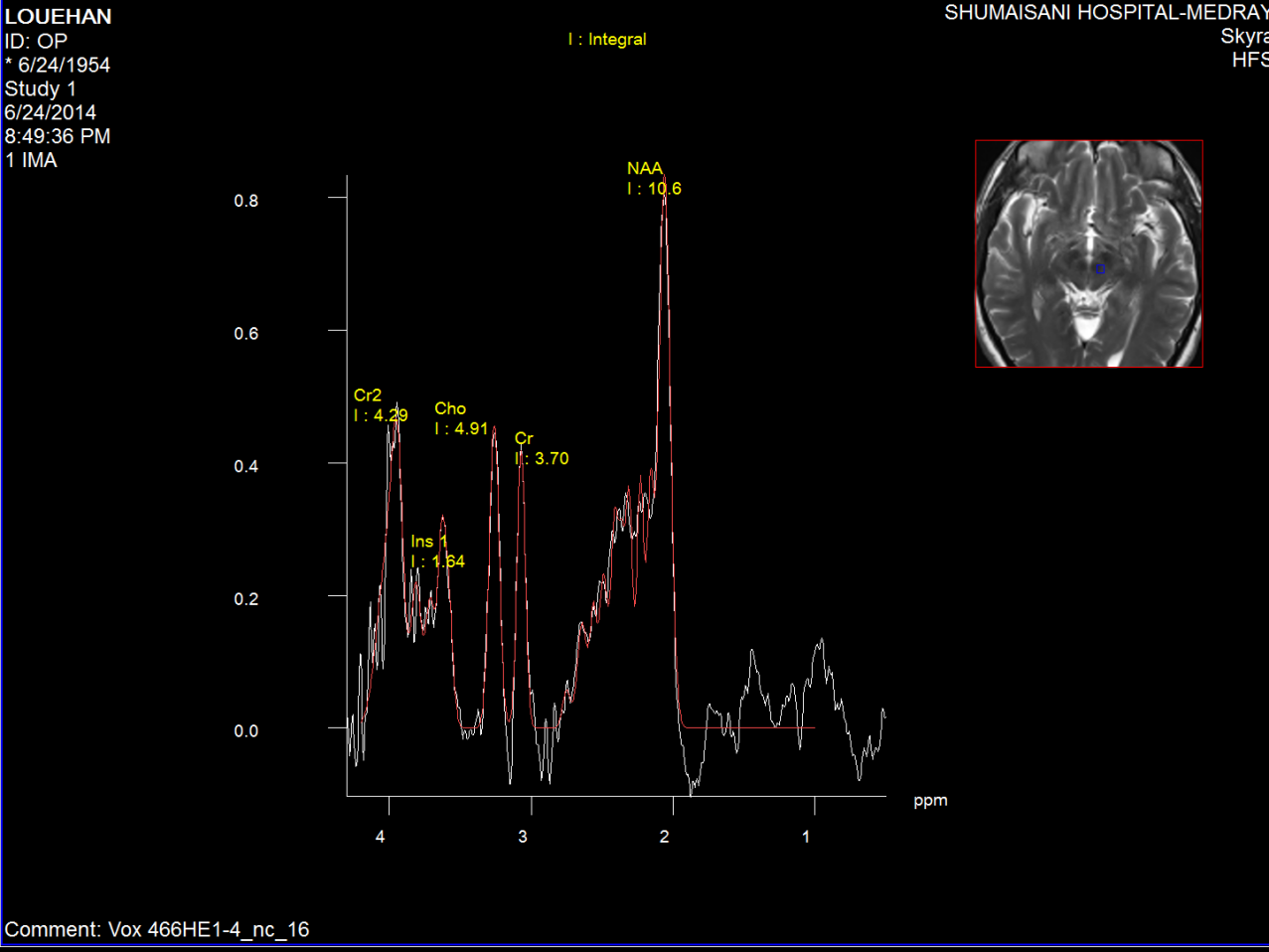
Figure-2: Left red nucleus short TE spectroscopy in
parkinsonian with tremor of the left side of the
body more the upper limb.
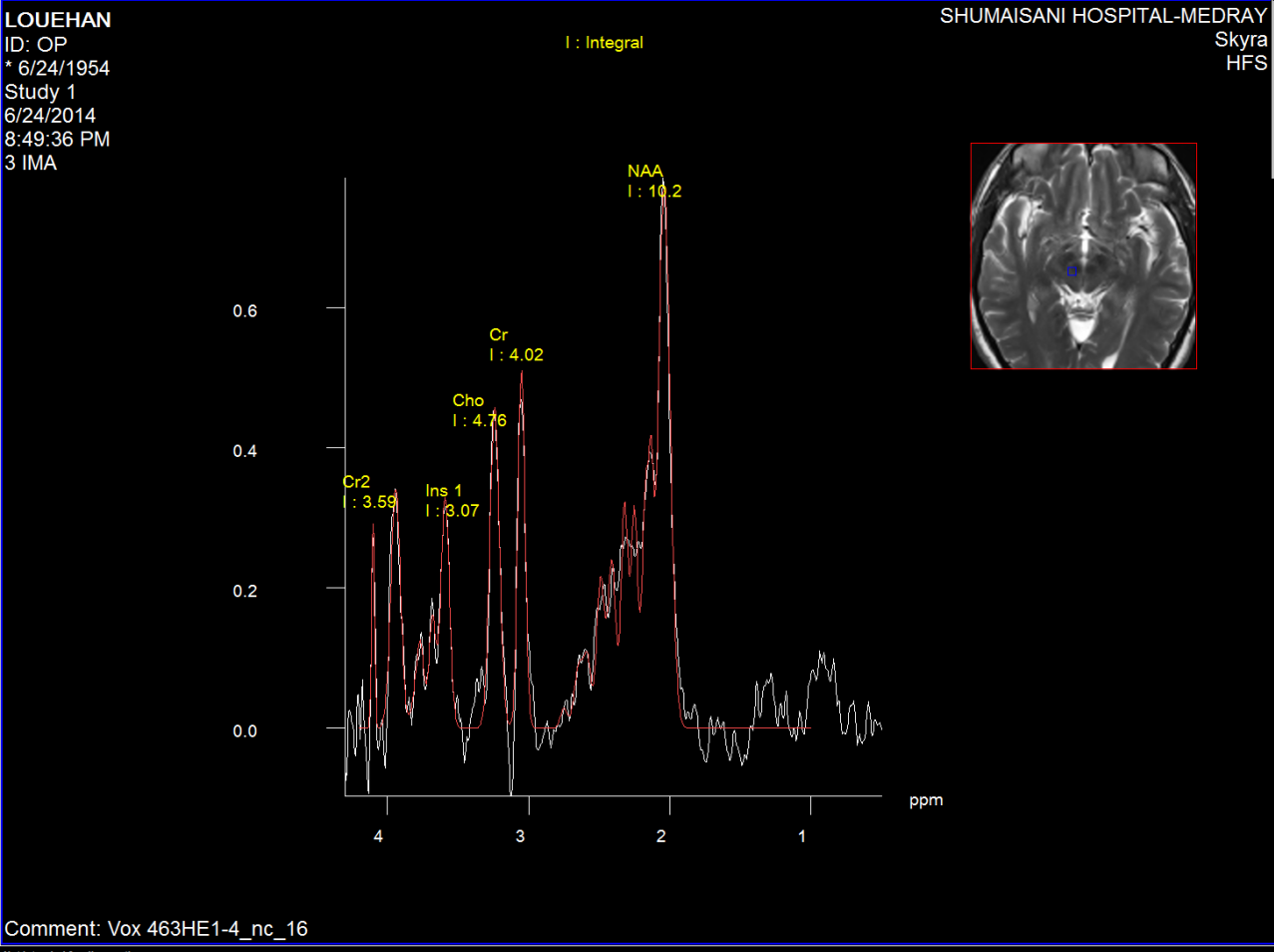
Figure-3: Short TE spectroscopy of the red nucleus
of the same patient in fig-2.
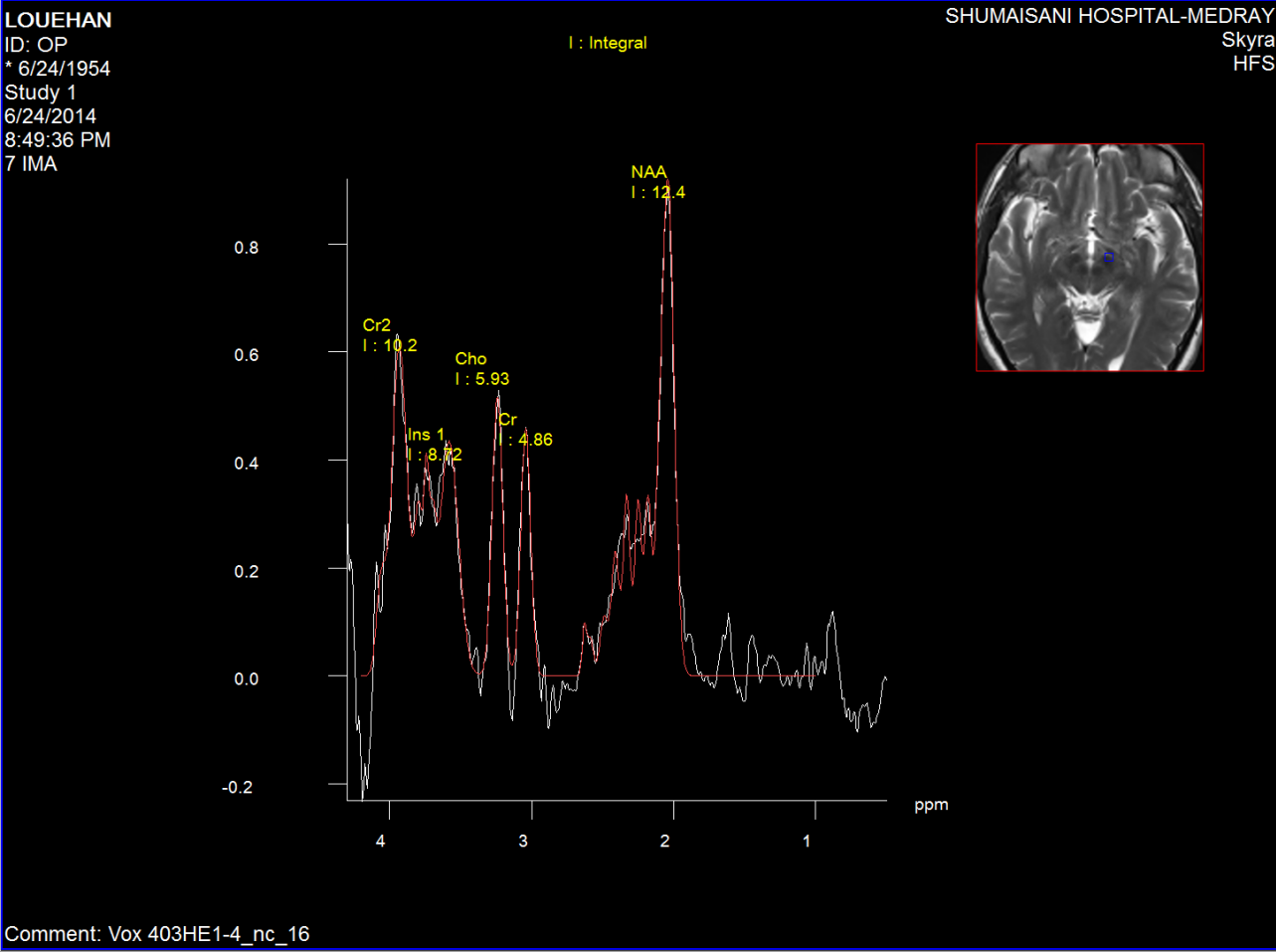
Figure-4: Short TE spectroscopy of the left
substancia nigra of the same patient as in
figure2,3.
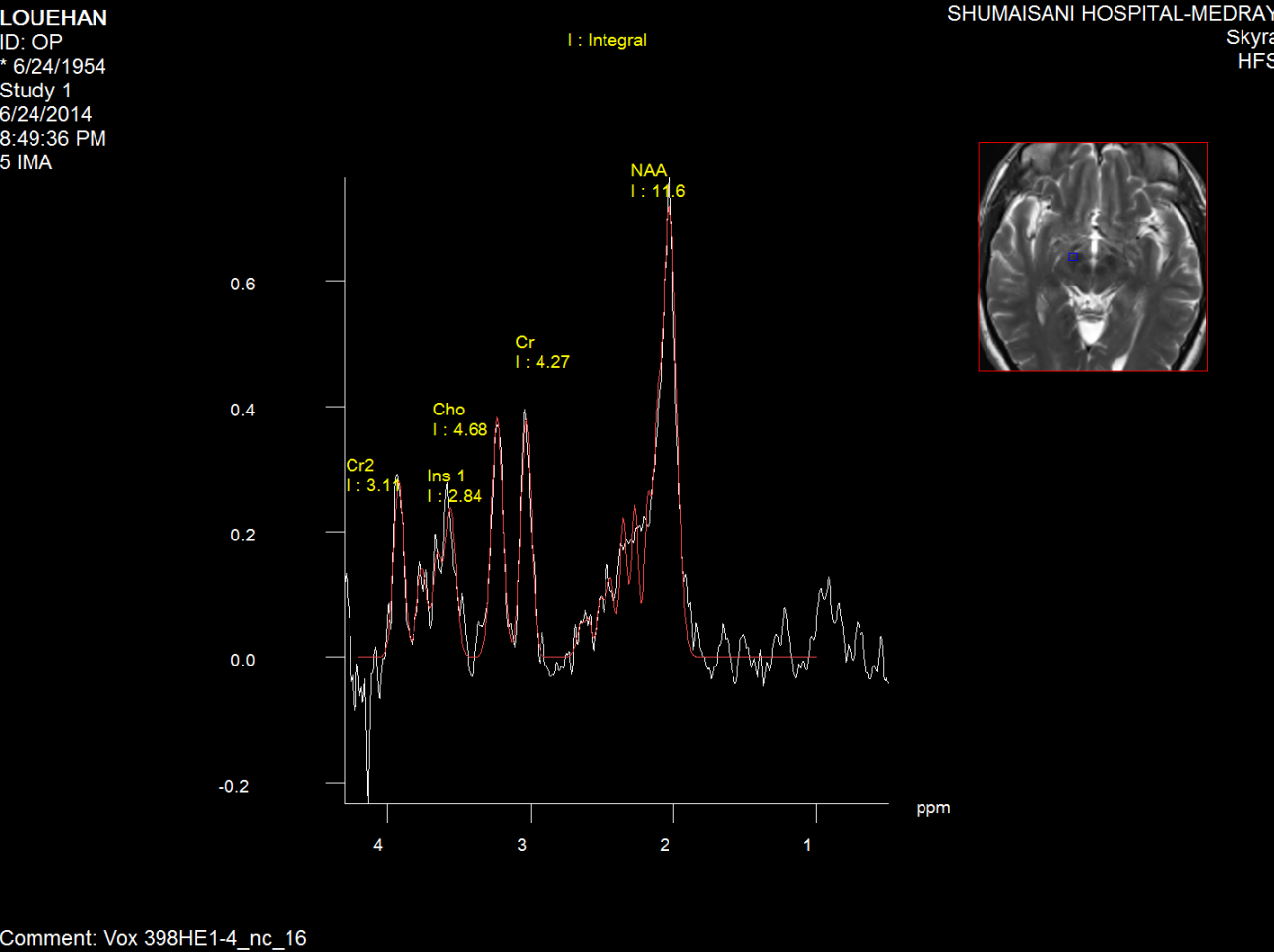
Fifure-5: Short TE spectroscopy of the right
substancia nigra of the same patient as in fig2.3.4.
References:
1. Oz G, Terpstra M, Tkac I, Aia P, Lowary J, Tuite
PJ, et al. Proton MRS of the unilateral substantia
nigra in the human brain at 4 tesla: Detection of
high GABA concentrations. Magn Reson Med 2006; 55:
296–301. |